S „nemendelovské dědictví„Máme na mysli jakýkoli vzor dědičnosti, kdy se zděděné znaky nerozdělují v souladu s ustanoveními Mendelových zákonů..
V roce 1865 provedl Gregor Mendel, považovaný za „otce genetiky“, řadu experimentálních křížení s rostlinami hrachu, jejichž výsledky ho vedly k navržení některých postulátů (Mendelovy zákony), které se snažily logicky vysvětlit dědictví. postavy mezi rodiči a dětmi.
Tento bystrý rakouský mnich pečlivě sledoval segregaci rodičovských genů a jejich vzhled u potomků jako dominantní a recesivní postavy. Kromě toho určil matematické vzorce popisující dědičnost z jedné generace na druhou a tato zjištění byla „uspořádána“ v podobě 3 základních zákonů:
- Zákon dominance
- Zákon o oddělení postav a
- Zákon nezávislé distribuce.
Mendelovy úspěchy a dedukce byly skryty po mnoho let až do jejich znovuobjevení na počátku 20. století..

V té době si však vědecká komunita zachovala poněkud skeptický postoj, pokud jde o tyto zákony, protože se nezdálo, že by vysvětlovaly vzory dědičnosti u žádného živočišného nebo rostlinného druhu, zejména u znaků určených více než jedním lokusem..
Z tohoto důvodu první genetici klasifikovali dědičné vzorce pozorované jako „Mendelian“ (ty, které lze vysvětlit oddělením jednoduchých, dominantních nebo recesivních alel patřících do stejného místa) a „non-Mendelian“ (ty, které lze snadno vysvětlit).
Rejstřík článků
Mendelianova dědičnost se týká dědičného vzorce, který je v souladu se zákony segregace a nezávislé distribuce, podle nichž gen zděděný od kteréhokoli z rodičů vylučuje v gametách se stejnou frekvencí nebo spíše se stejnou pravděpodobností.
Hlavní vzory Mendelovy dědičnosti, které byly popsány u některých onemocnění, jsou: autosomálně recesivní, autozomálně dominantní a spojené s chromozomem X, které se přidávají k vzorům dominance a recesivity popsaným Mendelem.
Ty však byly postulovány s ohledem na viditelné znaky, a ne na geny (je třeba poznamenat, že některé alely mohou kódovat znaky, které se segregují jako dominantní, zatímco jiné mohou kódovat stejné znaky, ale tyto se segregují jako recesivní geny).
Z výše uvedeného vyplývá, že nemendelovská dědičnost se skládá jednoduše z jakéhokoli dědičného vzoru, který nesplňuje normu, ve které se gen zděděný od kteréhokoli z rodičů vylučuje v zárodečných buňkách se stejnou pravděpodobností, a to zahrnuje:
- Mitochondriální dědičnost
- "Otisk"
- Uniparental disomy
- Neúplná dominance
- Kodominance
- Více alel
- Pleiotropie
- Smrtící alely
- Polygenní vlastnosti
- Dědičnost spojená se sexem
Výskyt těchto variací v dědičných vzorcích lze přičíst různým interakcím, které mají geny s jinými buněčnými složkami, kromě toho, že každý z nich podléhá regulaci a variaci v kterékoli z fází transkripce, sestřihu, translace, proteinu skládání, oligomerizace, translokace a kompartmentalizace v buňce a pro její export.
Jinými slovy, existuje řada epigenetických vlivů, které mohou upravit vzorce dědičnosti jakéhokoli znaku, což vede k „odchylce“ od Mendelových zákonů..
Mitochondriální DNA také přenáší informace z jedné generace na druhou, stejně jako ta, která je obsažena v jádru všech eukaryotických buněk. Genom kódovaný v této DNA zahrnuje geny nezbytné pro syntézu 13 polypeptidů, které jsou součástí podjednotek mitochondriálního respiračního řetězce, nezbytných pro organismy s aerobním metabolizmem..
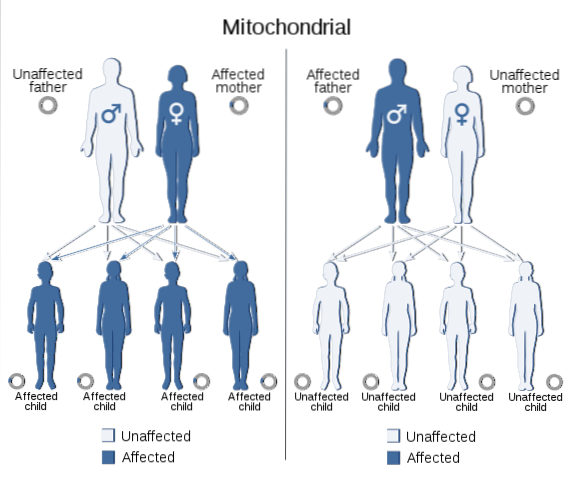
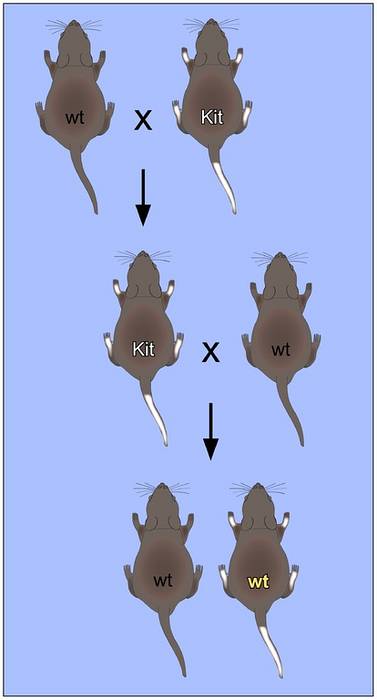
Ty rysy, které jsou výsledkem mutací v mitochondriálním genomu, vykazují specifický segregační vzorec, který se nazývá „mitochondriální dědičnost“, který se obvykle vyskytuje prostřednictvím mateřské linie, protože vajíčko poskytuje celkový doplněk mitochondriální DNA a spermie nepřispívají k žádným mitochondriím..
Genomický otisk se skládá z řady epigenetických „značek“, které charakterizují určité geny nebo úplné genomové oblasti a které jsou výsledkem genomického přechodu muže nebo ženy procesem gametogeneze..
Existují clustery otisků genů, které se skládají z 3 až 12 genů distribuovaných mezi 20 a 3700 kilobázemi DNA. Každý klastr má oblast známou jako kontrolní oblast imprintingu, která vykazuje specifické epigenetické modifikace od každého rodiče, včetně:
- Methylace DNA na specifických alelách v cytokinových zbytcích párů CpG
- Posttranslační modifikace histonů souvisejících s chromatinem (methylace, acetylace, fosforylace atd. Aminokyselinových zbytků těchto proteinů).
Oba typy „značek“ trvale modulují expresi genů, na kterých se nacházejí, a upravují tak své vzorce přenosu na další generaci..
Vzory dědičnosti, ve kterých exprese nemoci závisí na konkrétních alelách, které jsou zděděny od jednoho z rodičů, jsou známé jako efekt rodičovského původu..
Tento jev je výjimkou z Mendelova prvního zákona, který stanoví, že na potomky se přenáší pouze jedna ze dvou alel přítomných v každém z rodičů a podle chromozomálních zákonů dědičnosti lze přenášet pouze jeden z rodičovských homologních chromozomů na. další generace.
Toto je výjimka z pravidla, protože uniparental disomy je dědičnost obou kopií homologního chromozomu od jednoho z rodičů. Tento typ dědičného vzoru nemusí vždy vykazovat fenotypové vady, protože zachovává numerické a strukturní vlastnosti diploidních chromozomů..
Tento vzor dědičnosti sestává, fenotypicky vzato, ze směsi alel-kódovaných znaků, které jsou kombinovány. V případech neúplné dominance vykazují heterozygotní jedinci směs vlastností dvou alel, které je ovládají, což znamená, že vztah mezi fenotypy je upraven.
Popisuje dědičné vzorce, ve kterých jsou dvě alely, které jsou přenášeny z rodičů na jejich děti, současně vyjádřeny u těch s heterozygotním fenotypem, u nichž jsou obě považovány za „dominantní“.
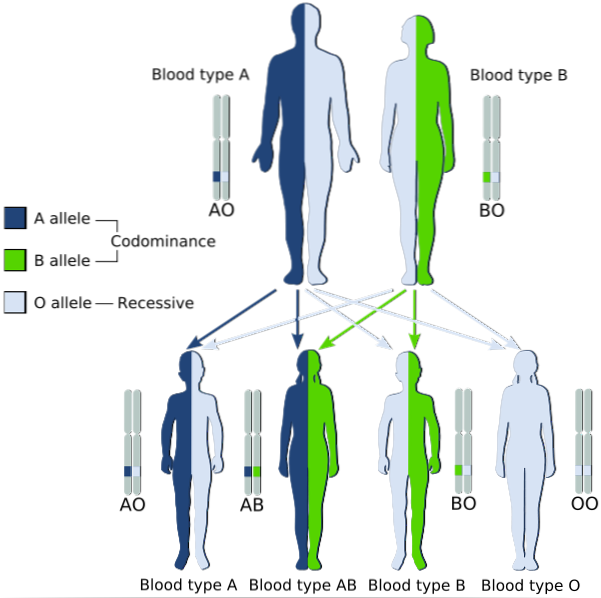
Jinými slovy, recesivní alela není „maskována“ expresí dominantní alely v alelickém páru, ale oba jsou exprimovány a ve fenotypu je pozorována směs těchto dvou znaků..
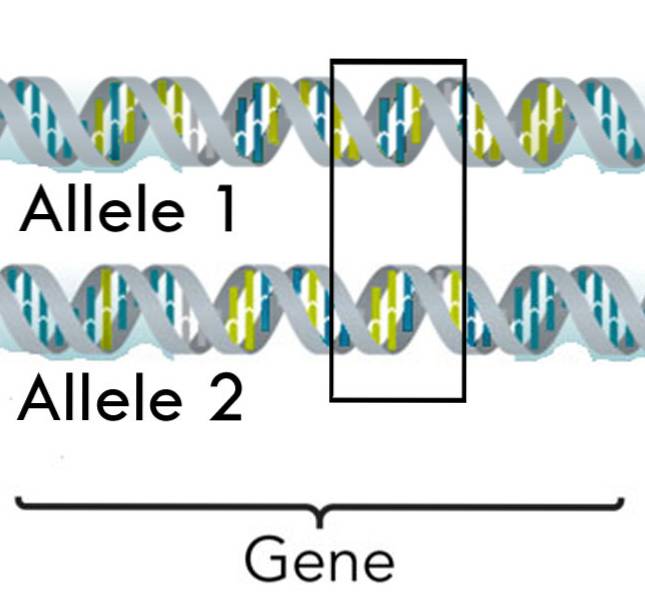
Možná jednu z hlavních slabin mendelovského dědictví představují rysy, které jsou kódovány více než jednou alelou, což je u lidí a mnoha dalších živých bytostí docela běžné..
Tento dědičný jev zvyšuje rozmanitost znaků, které jsou kódovány genem, a navíc tyto geny mohou kromě jednoduché nebo úplné dominance také zažít vzorce neúplné dominance a kodominance..
Další z „kamenů v botě“ nebo „uvolněných nohou“ Mendelovy dědičné teorie souvisí s těmi geny, které řídí vzhled více než jednoho viditelného fenotypu nebo charakteristiky, jako je tomu v případě pleiotropních genů..
Mendel ve svých pracích také neuvažoval o dědičnosti určitých alel, které by mohly zabránit přežití potomků, pokud jsou v homozygotní nebo heterozygotní formě; to jsou smrtící alely.
Letální alely obvykle souvisejí s mutacemi nebo defekty genů, které jsou nezbytně nutné pro přežití, které jsou při přenosu na další generaci (takové mutace) v závislosti na homozygositě nebo heterozygositě jednotlivců smrtelné.
Existují vlastnosti, které jsou kontrolovány více než jedním genem (s jejich alelami) a které jsou navíc silně kontrolovány prostředím. U lidí je to extrémně běžné a je tomu tak u vlastností, jako je výška, barva očí, vlasů a kůže, stejně jako u rizika utrpení některých nemocí.
U lidí a mnoha zvířat existují také rysy, které se nacházejí na jednom ze dvou pohlavních chromozomů a které se přenášejí sexuální reprodukcí. Mnoho z těchto rysů je považováno za „spojeno se sexem“, pokud jsou prokázány pouze u jednoho z pohlaví, ačkoli obě jsou fyzicky schopné tyto znaky zdědit..
Většina rysů souvisejících s pohlavím je spojena s některými recesivními chorobami a poruchami.
U lidí existuje genetická porucha známá jako Marfanův syndrom, která je způsobena mutací v jediném genu, který současně ovlivňuje růst a vývoj (mimo jiné výšku, vidění a funkci srdce)..
Toto je případ považovaný za vynikající příklad nemendelovského vzorce dědičnosti zvaného pleiotropie, ve kterém jediný gen ovládá několik charakteristik.
Genetické poruchy, které jsou výsledkem mutací v mitochondriální DNA, představují řadu klinických fenotypových variací, protože dochází k tomu, co je známé jako heteroplazmy, kde různé tkáně mají různé procento mutantního mitochondriálního genomu, a proto představují různé fenotypy.
Mezi tyto poruchy patří mitochondriální „depleční“ syndromy, což je skupina autosomálně recesivních poruch charakterizovaných významným snížením obsahu mitochondriální DNA, která končí systémy nedostatečné produkce energie v těch postižených orgánech a tkáních..
Tyto syndromy mohou být způsobeny mutacemi v jaderném genomu, které ovlivňují jaderné geny zapojené do syntézy mitochondriálních nukleotidů nebo do replikace mitochondriální DNA. Účinky lze prokázat jako myopatie, encefalopatie, hepato-cerebrální nebo neuro-gastrointestinální defekty..



Zatím žádné komentáře