The Canavanova choroba Jde o vzácné genetické onemocnění, ke kterému dochází proto, že nervové buňky v mozku jsou poškozené a nemohou spolu komunikovat. Toto onemocnění je přítomno v jakékoli společnosti a etnické skupině, i když je mnohem častější u aškenázské židovské populace a jejich potomků, kde je postižen 1 ze 6 400–13 000 lidí. Globální prevalence není známa.
Toto onemocnění patří do skupiny leukodystrofií. Tato kategorie zahrnuje všechny genetické poruchy, při kterých je myelinový obal obklopující axony neuronů poškozen, a proto mezi neurony není dobrá komunikace.
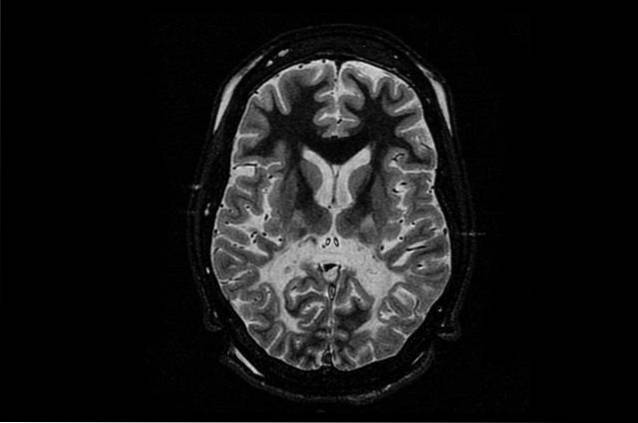
Nejběžnější a zároveň nejzávažnější formou tohoto onemocnění je novorozenec nebo dítě. Tato forma Canavanovy choroby postihuje novorozené děti nebo v jejich prvních letech života..
Děti, které trpí tímto onemocněním, nepředstavují během prvních měsíců života žádné problémy, ale začnou kvést mezi 3 a 5 měsíci. Hlavní příznaky jsou způsobeny deficitem vývoje, kdy děti mají motorické problémy, které jim brání v otáčení, otáčení hlavy nebo v sedění bez jakékoli podpory.
Dalšími běžnými příznaky jsou svalová slabost (hypotonie), abnormální vývoj hlavy (makrocefalie) a podrážděnost. V menší míře mohou mít také problémy s jídlem, záchvaty a problémy se spánkem..
Další méně častou formou je Canavanova choroba, která začíná ve středním dětství nebo dospívání. Děti a dospívající s tímto onemocněním mají problémy s vývojem jazyka a motorickými dovednostmi, ale tyto problémy jsou často tak mírné, že nejsou identifikovány jako příznaky Canavanovy choroby..
Očekávaná délka života lidí s Canavanovou chorobou je velmi heterogenní a liší se zejména v závislosti na době nástupu nemoci.
Děti, které trpí novorozeneckou nebo kojeneckou formou, obvykle žijí jen několik let, i když některé dosáhnou dospívání a velmi málo až do dospělosti. Zatímco ti, kteří trpí juvenilní formou, mají normální délku života.
Rejstřík článků
Existují dvě dobře diferencované formy Canavanovy choroby: forma novorozeneckého nebo kojeneckého nástupu a forma nástupu ve středním dětství nebo dospívání..
Příznaky novorozenecké nebo Canavanovy choroby začínající v dětství jsou velmi závažné, obvykle nejsou patrné do 3–50 měsíců věku a zahrnují makrocefalii, ztrátu motorické kontroly hlavy a vývojové deficity. Vývojové deficity se stávají zřetelnějšími, jak dítě stárne.
Nejzávažnějšími příznaky jsou příznaky spojené s motorickými problémy, protože děti nejsou schopny sedět nebo vstát bez podpory, chodit nebo mluvit. Když stárnou, hypotonie může vést ke spasticitě.
I když mají všechny tyto motorické problémy, mohou se naučit společensky komunikovat, usmívat se, ukazovat na předměty ...
Některé děti také trpí optickou atrofií, která způsobuje zrakové problémy, i když stále mohou vizuálně identifikovat objekty.
Jak příznaky rostou, zhoršují se a způsobují potíže se spánkem, záchvaty a potíže s krmením. Dítě se stává zcela závislým a potřebuje pomoc při provádění jakéhokoli úkolu.
Očekávaná délka života těchto dětí je poměrně krátká, většina z nich zemře za několik let, i když některé žijí až do dospívání nebo dospělosti.
Canavanova choroba s nástupem ve středním dětství nebo dospívání je mírnější než ta předchozí. Mezi příznaky patří určité potíže ve verbálním a motorickém vývoji.
I když jsou obvykle tak mírné, že nejsou identifikovány jako příznaky Canavanovy choroby, toto onemocnění je obvykle diagnostikováno po provedení analýzy moči, protože jedním ze znaků je vysoká koncentrace kyseliny N-acetyl asparagové (NAA v moči)..
Toto onemocnění je způsobeno mutací genu zvaného ASPA. Tento gen je ten, který řídí enzym aspartoacylázu, který je zodpovědný za degradaci molekul NAA..
Mutace genu ASPA způsobuje, že aspartoacyláza snižuje její účinnost, takže nedegraduje dostatek molekul NAA a bude zde vysoká koncentrace této látky. Čím dříve k této mutaci dojde, tím horší účinky má.
I když fungování molekul NAA není zcela dobře pochopeno, zdá se, že se podílejí na transportu molekul vody přes neurony a přebytek této látky brání tvorbě nového myelinu a ničí ten stávající. To způsobuje, že spojení mezi neurony nefunguje správně a mozek se nedokáže normálně vyvíjet..
Kromě toho může být tato nemoc zděděna autozomálně recesivně. Pokud je tedy každý člen páru nositelem patogenní varianty genu ASPA a rozhodne se mít dítě, je pravděpodobné, že:
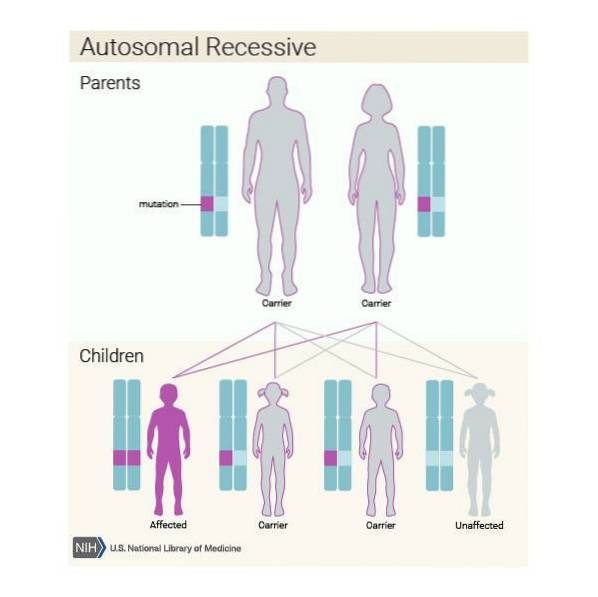
Je velmi důležité, aby jednotlivci patřící k rizikové populaci, v tomto případě potomci aškenázských Židů, podstoupili genetickou analýzu, aby zkontrolovali, zda jsou nositeli genu ASPA, než budou mít dítě..
Léčba závisí na formě onemocnění a na příznacích, které každý jedinec vykazuje..
V současné době neexistuje lék na Canavanovu chorobu, takže dostupné terapie se zaměřují na zlepšení kvality života pacienta podporou, výživou a hydratací a prevencí a léčbou infekcí.
Doporučuje se, aby děti dostaly fyzioterapeutickou léčbu ke zlepšení držení těla a motorických dovedností, aby se vyhýbaly a léčily kontraktury a svalové problémy, jako jsou dekubity. Mohou se také účastnit terapeutických a vzdělávacích programů ke zlepšení svých komunikačních dovedností..
Léčba léky zahrnuje antiepileptika (AED), pokud má dítě záchvaty, acetazolamid (značka Diamox®) ke snížení intrakraniálního tlaku a injekcí botulotoxinu (Botox®) k léčbě spasticity, pokud je přítomna.
Je nutné každých 6 měsíců provádět kontrolu, aby se zkontrolovalo, v jakém stavu se dítě nachází a jak probíhá jeho vývoj.
Lidé, kteří trpí touto formou onemocnění, pociťují mnohem mírnější příznaky, takže obvykle potřebují pouze terapie ke zlepšení jazyka nebo speciální vzdělávací programy. Nepotřebují žádné léky.
Doporučuje se každoroční sledování stavu dítěte.
Účinnost dalších terapií je v současné době studována na modelech u lidí i zvířat..
Účinnost genetické transplantace do mozku dětí s Canavanovou chorobou se zkoumá pomocí nevirového vektoru.
První výsledky ukazují, že tento typ transplantace je dobře snášen dětmi a způsobuje určité biochemické, radiologické a metabolické změny, ale pro léčbu nemoci není užitečný, proto se stále provádějí testy (Leone et al 2000, Janson et al. . až 2002).
McPhee a kol. (2006) provádějí studii, ve které je zdravý gen ASPA transplantován na různá místa v těle dětí pomocí AAV2 jako vektoru. V jednom z testů, kterého se zúčastnilo 10 dobrovolnických dětí. U 3 z nich transplantace fungovala a neutralizovala jejich protilátky, ale žádné z dětí se nezlepšilo.
Citrát lithný může snížit hladinu koncentrace NAA v mozku, a proto Assadi et al. (2010) se rozhodli provést experiment, při kterém podávali citrát lithný 6 lidem s Canavanovou chorobou po dobu 60 dnů.
Byly nalezeny hladiny koncentrace NAA v bazálních gangliích a bílé hmotě frontálního laloku, ačkoli nebyla nalezena žádná klinická zlepšení.
Nedostatek aspartoacylázových enzymů způsobuje nízké hladiny acetátu v mozku, proto se Mahavarao a jeho tým (2009) rozhodli podat glycerol triacetát dvěma pacientům s Canavalovou chorobou, aby zvýšili hladinu acetátu a zjistili, zda se tím zvýšila i hladina aspartoacylázy.
Sloučenina byla pacienty dobře tolerována, ačkoli nebyla nalezena žádná klinická zlepšení. V současné době provádějí testy podávání vyššího množství triacetátu glycerolu.
Jedním ze způsobů, jak vytvořit zvířecí modely, které představují nemoc, je vytvořit zvířata knokaut. Tato zvířata, obvykle myši, jsou geneticky modifikována tak, aby odstranila nebo změnila gen, který se při nemoci změnil. V tomto případě je modifikovaným genem gen ASPA..
Zvířecí modely se používají k lepšímu pochopení nemoci, studiu její biologické korelace a ověření účinnosti nové léčby.
Matalon a kol. (2003) použili myši knokaut testovat účinnost genové terapie s AAV2 jako vektorem. Zjistili, že došlo k vylepšení myelinových obalů, ale pouze v některých částech, ne v celém mozku.
Surendranův tým ve spolupráci s Genzyme Corporation (2004) testoval léčbu transplantací kmenových buněk. Zjistili, že byly vyrobeny nové oligodendrocyty, ale to nestačí na obnovení všech myelinových obalů..
Další tým testoval terapii, která spočívala v nahrazení nefunkčních asparthoacyklázových enzymů novými, které byly injikovány do pobřišnice myší. knokaut.
Krátkodobé výsledky ukázaly, že enzymy dokázaly projít hematoencefalickou bariérou (dosáhnout svého cíle) a dokázaly významně snížit hladinu NAA v mozku. I když jsou tyto výsledky slibné, je k ověření dlouhodobých účinků nutná longitudinální studie (Zano et al., 2011).
První příznaky, které upozorňují lékaře, že něco není v pořádku, jsou fyzické, zejména hypotonie a makrocefalie.
Za normálních okolností, pokud jsou tyto příznaky pozorovány, se u dítěte obvykle provádí neurouimagingová studie, aby se zkontrolovaly příznaky leukodystrofie, jako je nižší hustota bílé hmoty. Je třeba poznamenat, že tento test je méně účinný u dětí s Canavanovou chorobou, která začíná ve středním dětství nebo dospívání..
Jakmile se prokáže, že dítě trpí leukodystrofií, provedou se konkrétnější testy k vyloučení dalších nemocí, mezi ně patří:
Posledním krokem k potvrzení nemoci by bylo provést genetickou studii takto:



Zatím žádné komentáře