The phakomatosis je skupina neurokutánních poruch genetického původu, v běžné populaci vzácná. Na klinické úrovni jsou charakterizovány vývojem multisystémového organického postižení kůže nebo nádorových lézí v různých oblastech kůže, orgánů nebo nervového systému..
Jeho nespecifický klinický průběh navíc ztěžuje včasnou diagnostiku, takže jeho zdravotní a psychologické důsledky významně zhoršují kvalitu života postižené osoby a jejích příbuzných..
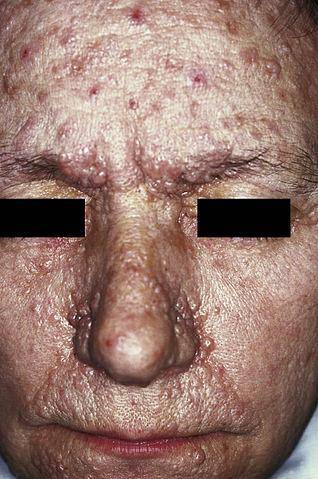
I když existuje velké množství neurokutánních onemocnění, mezi nejčastější patří fibromatóza typu I a typu II, Bournevilleova choroba, Sturge-Weberův syndrom a Von Hippel-Lindauova choroba..
Na druhou stranu, navzdory skutečnosti, že se jedná o vrozené patologie, bylo navrženo několik terapeutických přístupů dermatologické povahy, které se snaží zlepšit příznaky a příznaky charakteristické pro tyto poruchy, a tedy i lékařskou prognózu postižených.
Rejstřík článků
Termín phakomatosis pochází z výrazu řeckého původu Phakos jehož význam se týká <
Neurokutánní patologie jsou zásadně charakterizovány existencí významné souvislosti mezi neurologickým postižením nebo poruchou a dermatologickými projevy.
Termín neurokutánní patologie se tedy používá obecně, aby zahrnoval různá onemocnění, která jsou přítomna u vrozeně postižené osoby a která navíc mohou být přítomna po celý život s rozvojem kožních lézí a nádorů v různých oblastech, nervovém systému, kardiovaskulární systém, ledvinový systém, kožní systém, oční systém atd..
Tímto způsobem byl termín phakomatosis zaveden v roce 1917 Brouwerem a později van der Hoeve v roce 1923, počáteční popisy však odkazovaly pouze na některé patologie zahrnuté do této skupiny. V současné době více než 40.
Na klinické úrovni je fakomatóza popsána jako onemocnění, které se projevuje kožními změnami a benigními / maligními malformacemi v různých systémech: neurologické, oční, kožní a viscerální..
Pokud jde o postižené oblasti, různí autoři poukazují na to, že nejvíce zasaženy jsou oblasti ektodermálního původu, tj. Kůže a nervový systém, i když mohou ovlivnit i jiné systémy nebo zařízení, například oko.
Syndromy a patologie neurokutánního původu jsou v běžné populaci vzácnými chorobami, ačkoli o všech těchto údajích na obecné úrovni neexistují žádné specifické údaje.
Epidemiologie těchto poruch se tedy liší v závislosti na typu onemocnění, konkrétně neurofibromatóza je jednou z nejběžnějších, s relativní prevalencí jednoho případu na 300 000 porodů.
Neurokutánní onemocnění jsou charakterizována vývojem kožních lézí. Konkrétně se phakomatóza odlišuje od mnoha jiných přítomností hamartomů..
Hamartomy jsou druh benigní malformace nebo nádoru, který může růst v různých orgánech, jako je mozek, srdce, oči, kůže nebo plíce..
Fakomatóza však může být spojena s celou řadou zdravotních stavů, které se budou zásadně lišit v závislosti na konkrétním onemocnění nebo patologii postižené osoby..
V současné době bylo na klinické a genetické úrovni identifikováno velké množství neurokutánních poruch, existují však některé s vyšší prevalencí v běžné populaci: neurofibromatóza typu I a typu II, Bournevilleova choroba, Vonova choroba Hippel-Lindau a Sturge-Weber syndrom.
Existují různé klinické formy neurofibromatózy. V současnosti jsou však nejčastější neurofibromatóza typu I, nazývaná také Von Reclinghausenova choroba, a neurofibromatóza typu II, následovaná spinální shwannomatózou.
Na etiologické úrovni mají všechny tyto lékařské projevy neurofibromatózy genetický původ a vyskytují se při tvorbě nádorů v nervových oblastech, zejména v centrálním a periferním nervovém systému..
Nádorové formace, obvykle nerakovinné nebo benigní, mají tendenci růst a vyvíjet se téměř kdekoli v nervovém systému, jako je mozek, mícha nebo periferní nervy.
Řasy sekundárních zdravotních komplikací neurofibromatózy tedy zahrnují abnormality v růstu, rozvoj záchvatů, výskyt mozkových nádorů, kostní patologie, hluchota a / nebo slepota nebo vývoj významných problémů s učením, mimo jiné.
Tato patologie je navíc přítomna od okamžiku narození. Významný projev jeho klinického obrazu však může být odložen až do pozdního dětství, raného dospívání nebo dospělosti..
Na druhou stranu diagnóza tohoto typu patologie obvykle zahrnuje kromě fyzického a neurologického vyšetření také různé neuroimagingové testy a genetické analýzy..
Kromě toho v současné době neexistuje léčba neurofibromatózy, existují však specializované terapeutické přístupy v kontrole dermatologických postižení, mohou zahrnovat jak farmakologickou, tak chirurgickou léčbu k zastavení nebo eliminaci nádorových formací..
Neurofibromatóza typu I (NF1), známá také jako von Recklinghausenova choroba, se projevuje hlavně přítomností světle hnědých skvrn, běžně označovaných jako „café au lait“, ephelidů (pihy) a neurofibromů (poškození nervů ve Schwannových buňkách a neuritech).
Má autozomálně dominantní genetický původ, konkrétně kvůli mutaci na chromozomu 17, v místě 17q11.2. Gen zapojený do
vývoj neurofibromatózy typu I, má významnou roli v modulaci buněčného růstu a diferenciace a navíc může fungovat jako tumor supresor.
Pokud jde o epidemiologii této patologie, představuje přibližnou prevalenci jednoho případu na každých 2 500 300 porodů.
Diagnóza neurofibromatózy typu I je obvykle stanovena na základě konsensuálních klinických kritérií Národního zdravotního ústavu (1987), vyžaduje však nepřetržité sledování, aby se zabránilo sekundárním zdravotním komplikacím..
Obvykle jsou nádory léčeny léky, aby se zabránilo jejich exponenciálnímu vývoji nebo chirurgickým odstraněním.
Neurofibromatóza typu II (NF2) se projevuje hlavně vývojem schwannomů, tj. Nádorových útvarů odvozených od Shcwaanových buněk, které budou odpovědné za pokrytí prodloužení nervů.
Schwannomy nebo neuriomy obvykle postihují zejména sluchový a optický nerv a v menší míře oblasti kůže.
Neurofibromatóza typu II má autozomálně dominantní genetický původ, konkrétně kvůli přítomnosti mutace na chromozomu 22 v místě 22q11.22.
Gen podílející se na vývoji této patologie je zodpovědný za kódování proteinové složky s významnou rolí v potlačení nádoru, takže jeho nedostatečná aktivita produkuje abnormální zvýšení buněčné proliferace.
Pokud jde o epidemiologii této patologie, je méně častá než u typu 1, což představuje přibližnou prevalenci jednoho případu na 50 000 porodů.
Diagnóza neurofibromatózy typu II je podobná jako u předchozího typu a obvykle se stanoví na základě konsensuálních klinických kritérií Národního zdravotního ústavu. Obvykle však zahrnuje doplňkové laboratorní testy, jako je neuroimaging..
Obvykle se nádory léčí léky, avšak v případech, kdy je to možné, se používá chirurgické odstranění.
Bournevilleova choroba je jedním z termínů používaných k označení tuberózní sklerózy, genetické poruchy charakterizované přítomností hamartomů.
Na klinické úrovni může vést k multisystémovému ovlivnění charakterizovanému kožním ovlivněním (obličejové angiómy, fibromy nehtů, vláknité plaky, hypochromní skvrny atd.), Ovlivněním ledvin (renální angiomyolipomy nebo renální cysty), srdečním postižením (srdeční rhabdomyomy) , neurologické postižení (kortikální hlízy, subependymální gliové uzliny, atrocytomy, záchvaty, mentální postižení, chování a motorické abnormality), mimo jiné.
Stejně jako výše popsaná onemocnění je původ tuberózní sklerózy genetický. Konkrétně je to kvůli přítomnosti mutací v genech TSC1 a TSC2..
Na druhé straně je diagnóza tuberózní sklerózy stanovena na základě klinických kritérií navržených na lékařské konferenci v roce 1998. Genetická studie je však také považována za relevantní pro její potvrzení.
Pokud jde o léčbu tuberózní sklerózy, i když neexistuje léčba, obvykle se používají různé farmakologické a chirurgické přístupy, zejména pro kontrolu nádorových růstů a sekundárních zdravotních komplikací, jako jsou neurologické projevy.
Von Hippel-Lindauova choroba, známá také jako retino-cerebelární angiomatóza, se projevuje hlavně přítomností a vývojem vaskulárních malformací, cyst a / nebo nádorů, obecně benigních..
Má autozomálně dominantní genetický původ, konkrétně kvůli mutaci na chromozomu 3, v poloze 3p-25-26. Kromě toho představuje odhadovaný výskyt jednoho případu na 40 000 porodů..
Konkrétně Von Hippel-Lindauova choroba postihuje hlavně centrální nervový systém (CNS) a sítnici tvorbou hemangiomů.
Hemangiomy jsou vaskulární malformace charakterizované přítomností shluků rozšířených krevních kapilár. Obvykle se objevují v oblastech mozku a páteře, i když jsou také časté v sítnici nebo na kůži.
Diagnóza této patologie vyžaduje kromě fyzického a neurologického vyšetření také podrobnou oftalmologickou studii spolu s analýzou různých neuroimagingových testů, aby se potvrdila přítomnost nervových lézí..
Pokud jde o léčbu Von Hippel-Lindauovy choroby, základním zákrokem je chirurgický zákrok k odstranění vaskulárních malformací. Vyžaduje však nepřetržité sledování, aby se zabránilo sekundárním komplikacím..
Kromě toho má sníženou průměrnou délku života kolem 50 let, zejména v důsledku vývoje karcinomů renálních buněk (neoplastické formace rakovinných buněk v renálních tubulech).
Sturge-Weberův syndrom, známý také jako encefalo-trigeminální angiomatóza, se projevuje hlavně přítomností hemangiomů.
Hemangiom je typ novotvaru nebo tvorby nádoru charakterizovaný přítomností abnormálně vysokého počtu krevních cév v kůži nebo jiných vnitřních orgánech..
Konkrétně na klinické úrovni je Sturge-Weberův syndrom charakterizován vývojem hemangiomů obličeje, intrakraniálních hemangiomů a chorangických, spojivkových, epizcerálních a glaukomových hemangiomů..
Má genetický původ, konkrétně kvůli mutaci na chromozomu 9 v místě 9q21 v genu GNQ. Tato genetická složka má významnou roli v kontrole růstových faktorů, vazoaktivních peptidů a neurotransmiterů (Orhphanet, 2014).
Diagnóza Sturge-Weberova syndromu je stanovena na základě klinického podezření a různých laboratorních testů, jako je počítačová tomografie nebo magnetická rezonance..
Na druhou stranu, pokud jde o léčbu, laserová terapie je schopna snížit progresi této patologie a navíc v mnoha případech zcela eliminovat hemangiomy..


Zatím žádné komentáře