
The Apertův syndrom nebo akrocefalosyndaktylie typu I (ACS1) je patologie genetického původu, která je charakterizována přítomností různých alterací a malformací v lebce, obličeji a končetinách.
Na klinické úrovni je Apertův syndrom charakterizován přítomností nebo vývojem špičaté nebo protáhlé lebky, propadlé oblasti obličeje se změnou projekce zubů, fúzí a uzavřením kostí a kloubů prstů, proměnnou mentální retardací, poruchami jazyka , atd..
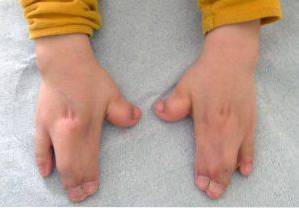
Ačkoli tato patologie může být dědičná, ve většině případů se Apertův syndrom vyskytuje bez přítomnosti rodinné anamnézy, v zásadě kvůli de novo mutaci během fáze těhotenství..
Genetické mechanismy, které způsobují Apertův syndrom, nejsou přesně známy. V současné době byly identifikovány různé genetické změny, které jsou schopné produkovat tuto patologii, v zásadě související s mutacemi v genu FGFR2.
Na druhé straně diagnóza Apertova syndromu obvykle začíná klinickým podezřením v prenatálním období po identifikaci abnormalit v rutinních ultrazvukových skenech a je potvrzena provedením genetické studie.
Pokud jde o léčbu, neexistuje žádný typ léčebného zásahu pro Apertův syndrom. V průběhu historie této patologie však byly navrženy různé specifické intervence, které obvykle zahrnují mimo jiné neurochirurgii, kraniofaciální chirurgii, maxilofaciální chirurgii, léčbu drogami, fyzikální terapii, psychologické a neuropsychologické intervence..
Rejstřík článků
Apertův syndrom je genetická patologie charakterizovaná přítomností různých kostních malformací na lebeční, obličejové a / nebo končetinové úrovni.
Podstatnou změnu Apertova syndromu představuje předčasné nebo předčasné uzavření lebečních trhlin, které způsobí abnormální růst zbytku struktur obličeje a lebky. Kromě toho se mohou objevit malformace také na horních a dolních končetinách, jako je fúze prstů na rukou a nohou..
Na druhé straně mohou být ovlivněny také kognitivní schopnosti lidí trpících Apertovým syndromem, s různou závažností od mírné po střední.
Ačkoli Baumgartner (1842) a Wheaton (1894) učinili první zmínky o tomto zdravotním stavu, nebylo to až do roku 1906, kdy francouzský lékařský specialista Eugene Apert přesně popsal tento syndrom a publikoval první klinickou zprávu.
Ve své publikaci Eugene Apert popisuje soubor nových případů pacientů postižených dobře definovaným malformativním vzorem a charakterizovaných charakteristickými znaky a příznaky této patologie..
Etiologické genetické faktory Apertova syndromu byly identifikovány až v roce 1995. Konkrétně Wilkie et al. Popsali přítomnost dvou mutací v genu FGFR2 u přibližně 40 postižených pacientů..
Apertův syndrom je navíc zdravotní stav, který je klasifikován jako onemocnění nebo patologie charakterizované přítomností kraniosynostózy (předčasné uzavření lebečních stehů).
Mezi další patologie patřící do této skupiny patří Pfeifferův syndrom, Crouzonův syndrom, Saethre-Chotzcenův syndrom a Carpenterův syndrom..
Apertův syndrom je považován za vzácnou nebo občasnou patologii, to znamená, že má prevalenci méně než jeden případ na 15 000 obyvatel běžné populace..
Konkrétně se Apertův syndrom vyskytuje u jedné osoby na každých 160 000–200 000 porodů a navíc existuje 50% pravděpodobnost přenosu této patologie na dědičné úrovni.
Navíc, pokud jde o rozdělení podle pohlaví, nebyla zjištěna vyšší prevalence u mužů nebo žen, ani nebyla spojena s konkrétními etnickými skupinami nebo geografickými lokalitami..
V současné době a vzhledem k tomu, že Apertův syndrom byl identifikován přibližně v roce 1984, v klinických zprávách a v lékařské literatuře, které publikovaly více než 300 případů této patologie.
Klinické projevy Apertova syndromu obvykle zahrnují malformaci nebo neúplný vývoj lebeční struktury, atypický fenotyp nebo obličejový obraz a změny skeletu v končetinách..
V případě Apertova syndromu souvisí centrální postižení s tvorbou a uzavřením kostní struktury lebky. Během embryonálního vývoje dochází k procesu zvanému creneosynostóza, který je charakterizován předčasným uzavřením lebečních stehů.
Kraniální trhliny nebo stehy jsou typem pásů vláknité tkáně, které mají základní cíl spojovat kosti tvořící lebku (čelní, týlní, temenní a temporální).
Během fáze těhotenství a raného postnatálního období je kostní struktura, která tvoří lebku, držena pohromadě díky těmto vláknitým a elastickým tkáním.
Za normálních okolností se lebeční kosti nespojí dříve než kolem 12 až 18 měsíců. Přítomnost měkkých míst nebo mezer mezi lebečními kostmi je součástí normálního vývoje dítěte.
Proto během celé fáze dětství umožňují tyto stehy nebo pružné oblasti mozku zrychleným růstem a navíc ho chrání před nárazy.
U Apertova syndromu tedy předčasné uzavření těchto lebečních stehů a lebečních kostí znemožňuje normální vývoj lebečního a mozkového růstu..
V důsledku toho mohou nejčastější příznaky a příznaky Apertova syndromu zahrnovat:
Tyto typy změn ovlivňují hlavně horní a dolní končetiny, obvykle fúzi a vývoj prstů..
Kromě toho je také možné pozorovat další klinické nálezy na úrovni pohybového aparátu, zkrácení různých kostí (rádius, pažní, stehenní), hypoplázie lopatky nebo pánve, fúze krčních obratlů.
V důsledku toho bude mít mnoho postižených sníženou pohyblivost kloubů, a proto se mohou vyvinout různé obtíže při získávání hrubé a jemné motoriky.
Tyto typy anomálií jsou mezi postiženými jedinci velmi heterogenní a variabilní, byly však identifikovány některé z nejčastějších:
Etiologická změna této patologie může vést k rozvoji lézí nebo sekundárních patologií na morfologické a strukturální úrovni v různých oblastech těla, mezi něž patří:
Navzdory skutečnosti, že v mnoha případech lze pozorovat přítomnost obecné změny kognitivních funkcí a intelektuální úrovně, mentální retardace není u všech případů Apertova syndromu jednoznačně přítomna..
Navíc v případech, kdy dojde k narušení intelektuální úrovně, může to být variabilní na stupnici od mírné po střední.
Na druhé straně v jazykové oblasti je častý vývoj různých deficitů, zejména v souvislosti s artikulací zvuků v důsledku mandibulárních a orálních malformací..
Apertův syndrom je způsoben přítomností specifické mutace v genu FGFR2. Experimentální studie ukázaly, že tento gen je zodpovědný za produkci proteinu nazývaného receptor 2 fibroblastového růstového faktoru.
Mezi funkcemi tohoto faktoru popisuje odesílání různých chemických signálů nezralým buňkám, aby došlo k jejich transformaci a diferenciaci na kostní buňky během fetální nebo prenatální fáze vývoje..
Přítomnost mutací v genu FGFR2 proto mění fungování tohoto proteinu, a proto může způsobit časnou fúzi kostí lebky, ruky a nohou..
Značnou část klinických rysů Apertova syndromu lze identifikovat během těhotenství, zejména při ultrazvukových vyšetřeních těhotenství a vývoje plodu..
Pokud tedy existuje klinické podezření, je znovu zahájena genetická studie k identifikaci přítomnosti genetické mutace kompatibilní s Apertovým syndromem..
Na druhou stranu, pokud jsou příznaky jemné nebo nebyly identifikovány před narozením, je možné provést podrobnou fyzikální analýzu a různé genetické testy k potvrzení diagnózy..
Ačkoli neexistuje žádný konkrétní lék na Apertův syndrom, byly popsány různé přístupy k léčbě příznaků a zdravotních komplikací této patologie..
Nejúčinnější terapeutické intervence jsou ty, které jsou implementovány brzy, v prvních okamžicích života a zahrnují profesionály z různých oblastí.
Léčba postižených dětí obvykle vyžaduje individuální plánování s plánovanými několika operacemi. Řízení této patologie je tedy založeno na nápravě malformací skeletu a kranio-obličeje a na psychologické a neuropsychologické podpoře..
Prostřednictvím neurochirurgie je cílem rekonstruovat lebeční klenbu, zatímco odborníci na maxilofaciální chirurgii se snaží napravit malformace obličeje. Na druhé straně je častá také účast úrazových chirurgů pro rekonstrukci malformací přítomných v rukou a nohou.
Kromě toho je přínosem pro dosažení optimálního, funkčního a nezávislého rozvoje postižených jedinců návrh individualizovaných programů pro včasnou stimulaci, komunikační rehabilitaci, nácvik sociálních dovedností nebo psychopedagogické sledování..



Zatím žádné komentáře