
The Joubertův syndrom je porucha genetického původu, která se vyznačuje snížením svalového tonusu, problémy s koordinací, abnormálními pohyby očí, změnami dýchacích vzorců a mentálním postižením (Joubert Syndrome Foundation, 2016).
Všechny tyto změny jsou způsobeny autosomálním genetickým přenosem, který způsobí důležité mozkové abnormality, snížení mozečkové vermis, jakož i abnormality ve struktuře mozkového kmene (National Institute of Neurological Disorders and Stroke, 2016).
Kromě toho je Joubertův syndrom součástí skupiny poruch nazývaných ciliopatie, které zahrnují dysfunkci části buněk zvaných řasinky. Joubert Syndrome Foundation, 2016).
Počáteční popis této patologie vytvořila Marie Joubert a kol. V roce 1968 byly popsány čtyři případy. U pacientů byla částečná nebo úplná absence mozečkové vermis, novorozenecký epizodický syndrom ampnea-hypernea, abnormální pohyby očí, ataxie a mentální retardace (Angemi a Zucotti, 2012).
Tento syndrom byl navíc spojen s různými multiorgánovými změnami, jako je jaterní fibróza, polydaktylie, nefronoptýza nebo retinální dystrofie (Angemi a Zucotti, 2012)..
Pokud jde o léčbu, v současné době neexistuje žádný lék na Joubertův syndrom. Terapeutické intervence jsou zaměřeny na symptomatickou kontrolu a podporu, fyzickou a intelektuální stimulaci dětí a pracovní terapii (National Institute of Neurological Disorders and Stroke, 2016).
Rejstřík článků
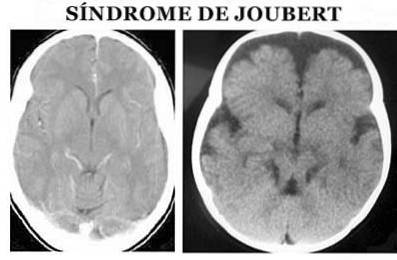
Joubertův syndrom (JS) je typ patologie genetického původu, který je charakterizován vrozenou malformací v oblastech mozkového kmene a agenezí (částečná nebo úplná absence) nebo hypoplázií (neúplný vývoj) cerebelární vermis, která může způsobit (Ophatnet , 2009).
Přesněji řečeno, na anatomické úrovni je charakterizován takzvaným molárním znakem středního mozku: ageneze nebo hypoplázie cerebelární vermis, zúžení horních mozečkových stopek se zesílením, prodloužením a nedostatkem dekubace a hlubokou interpedunkulární fossou Zuccoti, 2012).
Jedná se o poruchu, která může ovlivnit mnoho oblastí a orgánů těla, takže její příznaky se u postižených lidí značně liší (US National Library of Medicine, 2011).
Většina postižených trpí oslabeným svalovým tónem (hypotonie) a poruchami motorické koordinace (Ataxie). Mezi další charakteristické rysy patří: epizody změněného dýchání, nystagmus (nedobrovolný a arytmický pohyb očí), opožděný motorický vývoj a různé intelektuální potíže (US National Library of Medicine, 2011).
Prevalence Joubertova syndromu se odhaduje na přibližně 1/80 000 až 1/100 000 000 živě narozených dětí. Po celém světě bylo zaregistrováno více než 200 klinických případů (Angemi a Zuccoti, 2012).
Mnoho odborníků považuje tato čísla za podceňovaná, protože Joubertův syndrom má širokou škálu ovlivnění a je široce nediagnostikovaný (US National Library of Medicine, 2011).
Většina klinických příznaků Joubertova syndromu je v dětství více než evidentní, mnoho postižených dětí má výrazné motorické zpoždění (National Organization for Rare Disease, 2011).
Nejběžnějšími charakteristikami klinického průběhu jsou: nedostatek svalové kontroly (ataxie), změněné dechové vzorce (hyperkapnie), spánková apnoe, abnormální pohyby očí (nystagmus) a nízký svalový tonus (Národní organizace pro vzácná onemocnění, 2011).
Na druhou stranu některé změny, které mohou souviset s Joubertovým syndromem, zahrnují: změněný vývoj sítnice, abnormality duhovky, strabismus, změny ledvin a / nebo jater, výčnělek membrán pokrývajících mozek, mimo jiné ( Národní organizace pro vzácná onemocnění, 2011).
Všechny změny odvozené od tohoto syndromu jsou zahrnuty v několika oblastech: neurologické, oční, renální a muskuloskeletální změny (Bracanti et al., 2010).
Nejcharakterističtějšími neurologickými změnami Joubertova syndromu jsou Bracanti et al., 2010): hypotonie, ataxie, generalizované zpoždění vývoje, intelektuální změny, změny dýchacích cest a abnormální pohyby očí.
Na fyzické úrovni je sítnice jedním z orgánů postižených Joubertovým syndromem. Změny v tomto orgánu se objevují ve formě retinální dystrofie v důsledku progresivní degenerace buněk odpovědných za příjem fotografií.
Klinicky se oční změny mohou pohybovat od vrozené slepoty sítnice po progresivní degeneraci sítnice.
Na druhou stranu je také možné pozorovat přítomnost colobomu. Tato oční změna je vrozená vada, která postihuje oční duhovku a objevuje se jako díra nebo štěrbina.
Patologie související s funkcí ledvin postihují více než 25% pacientů postižených Joubertovým syndromem.
V mnoha případech mohou abnormality ledvin zůstat asymptomatické po několik let nebo se mohou začít projevovat nespecifickými příznaky, dokud se neprojeví jako akutní nebo chronické selhání ledvin..
Z prvních popisů této patologie je častým klinickým nálezem polydactialia (genetická porucha, která zvyšuje počet prstů na rukou nebo nohou)..
Kromě toho je také běžné pozorovat orofaciální nebo strukturní anomálie na úrovni páteře.
Experimentální studie klasifikovaly Joubertův syndrom jako autozomálně recesivní poruchu (National Organization for Rare Disease, 2011).
Autosomálně recesivní genetická porucha znamená, že pro přítomnost znaku nebo nemoci musí být přítomny dvě kopie abnormálního genu (National Institutes of Health, 2014).
Proto dochází k recesivní genetické změně, když osoba zdědí stejný abnormální gen pro stejnou vlastnost od každého rodiče. Pokud jedinec obdrží pouze jednu kopii genu souvisejícího s onemocněním, bude nosičem, ale nebude vykazovat příznaky (Národní organizace pro vzácná onemocnění, 2011).
Kromě toho bylo identifikováno nejméně deset genů jako jedna z možných příčin Joubertova syndromu (National Organization for Rare Disease, 2011).
Mutace v genu AHI1 je zodpovědná za tento patologický stav přibližně u 11% postižených rodin. U lidí s touto genetickou změnou jsou změny vidění běžné v důsledku vývoje retinální dystrofie (Národní organizace pro vzácná onemocnění, 2011)..
Mutace genu nphp1 je příčinou přibližně 1–2% případů Joubertova syndromu. U jedinců s touto genetickou alterací jsou alterace ledvin běžné (National Organization for Rare Disease, 2011).
Na druhou stranu je mutace genu CEP290 příčinou 4–10% případů Jouberova syndromu (National Organization for Rare Disease, 2011).
Kromě toho mutace v genech TME67, JBTS1, JBTS2, JBTS7, JBTS8 a JBTS9 také souvisí s vývojem Joubertova syndromu (National Organization for Rare Disease, 2011).
Diagnóza Joubertova syndromu je stanovena na základě fyzických příznaků. Je nutné provést jak podrobné fyzické vyšetření, tak i použití různých diagnostických testů, zejména obrazů magnetické rezonance (Ophatnet, 2009).
Kromě toho se také často používají molekulární genetické testy k identifikaci genetických změn, které byly prokázány u 40% případů Jouberova syndromu (National Organization for Rare Disease, 2011).
Na druhou stranu je také možné provést prenatální diagnostiku této patologie pomocí fetálního ultrazvuku a molekulární analýzy, zejména v rodinách s genetickou anamnézou Jouberova syndromu (Ophatnet, 2009).
Pokud se nejcharakterističtější rysy Joubertova syndromu vyskytnou v kombinaci s jednou nebo více dalšími fyzickými patologiemi, může být stanovena diagnóza Joubertova syndromu a souvisejících poruch (JSRD) (US National Library of Medicine, 2011).
Proto v závislosti na typu související patologie spojené s přítomností Joubertova syndromu můžeme najít jeho podtypy. Systém klasifikace Joubertova syndromu je však stále ve fázi evoluce díky objevu genetických příspěvků a lepší znalosti fenotypových korelací..
Můžeme tedy najít (Bracanti et al., 2010):
Léčba použitá při Joubertově syndromu je symptomatická a podporuje základní patologie. Kromě farmakologických intervencí je běžné používat časnou fyzickou a kognitivní stimulaci (National Institute of Neurological Disorders and Stoke, 2016).
Pokud jsou významné respirační změny, zejména v počátečních fázích života, je nutné sledovat respirační funkce (National Institute of Neurological Disorders and Stoke, 2016).
Na druhé straně by měla být identifikace a kontrola oční degenerace, ledvinových komplikací a ostatních komplikací souvisejících s Joubertovým syndromem provedena co nejdříve, aby bylo možné upravit terapeutická opatření (National Institute of Neurological Disorders and Stoke, 2016 ).


Zatím žádné komentáře