
The Ohtaharův syndrom, Také známá jako epileptická encefalopatie v raném dětství, jedná se o typ epilepsie charakterizované křečemi, záchvaty rezistentními na terapeutické přístupy a těžkou psychomotorickou retardací. Tento typ epilepsie se vyznačuje tím, že je jednou z prvních, objevuje se během prvních měsíců života a je také jednou z nejméně častých.
Na etiologické úrovni může být tato patologie způsobena různými událostmi, včetně krvácení, infarktu, asfyxie nebo strukturálních změn na úrovni mozku. Ve více než 60% případů však nelze určit konkrétní příčinu.

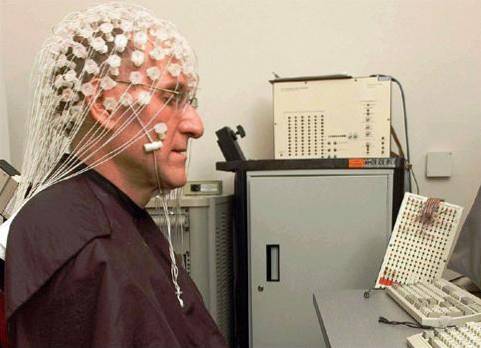
Pokud jde o diagnózu, za přítomnosti záchvatů a klinického podezření na epilepsii se obvykle používají různé diagnostické testy, jako je počítačová axiální tomografie (CT) nebo elektroencefalografie (EEG)..
Na druhou stranu, pokud jde o léčbu, různé přístupy obvykle nemají pozitivní výsledky, obvykle se používají dávky vitaminu B1, kyseliny valproové, vigabatrinu, ketogenní stravy atd..
Obecně platí, že děti s Ohtaharovým syndromem mívají špatnou lékařskou prognózu a umírají v krátkém časovém období. Existují však případy, kdy přežijí a postupují k Westovu syndromu..
Rejstřík článků
Ohtaharův syndrom je typ epileptické encefalopatie, různého původu a závislý na věku, s prvními klinickými projevy v prenatálním období.
Epilepsie je typ neurologické patologie, která postihuje hlavně centrální nervový systém. Ve většině případů jde o onemocnění s chronickým průběhem charakterizovaným rozvojem záchvatů nebo epileptických záchvatů..
Tyto události, které jsou výsledkem abnormální mozkové činnosti, jsou charakterizovány obdobími neobvyklých pocitů a chování, svalovými křečemi, chováním, dokonce ztrátou vědomí..
Epilepsie je dále považována za jednu z nejčastějších neurologických poruch na celém světě. Asi 50 milionů lidí trpí na celém světě epilepsií, avšak syndrom Ohtahara nebo dětská epileptická encefalopatie je onemocnění s nízkou prevalencí v běžné populaci.
V případě této patologie se termín encefalopatie používá konkrétně k označení různých poruch, které mění funkci a strukturu mozku.
Někteří autoři, jako Aviña Fierro a Herández Aviña, definují epileptickou encefalopatii jako soubor syndromů těžkých paroxysmálních záchvatů, které obvykle začínají klinicky v prvních okamžicích života nebo v raném dětství a které směřují k neodolatelné epilepsii, která se rychle vyvíjí směrem k smrt postižené osoby.
V roce 1976 tedy Ohtahara a jeho pracovní skupina popsali typ epileptické encefalopatie s časným nástupem a související s dalšími syndromy, jako je Lennox-Gastautův a Westův syndrom..
Podobně Clark v roce 1987 analýzou 11 případů potvrdil charakteristiky tohoto onemocnění a nazval jej Ohtaharův syndrom. Tímto způsobem byl Westův syndrom definován prostřednictvím následujících charakteristik:
Nakonec to nebylo až do roku 2001, kdy Mezinárodní liga proti epilepsii zahrnovala Ohtaharův syndrom jako konkrétní lékařskou entitu klasifikovanou v rámci epileptických encefalopatií, které se vyskytují u dětí..
Epilepsie je jednou z nejčastějších neurologických patologií, přibližně 50 milionů lidí postižených po celém světě (Světová zdravotnická organizace, 2016). Konkrétně různé studie odhadují její prevalenci na přibližně 4–10 případů na 1 000 obyvatel..
Ohtaharův syndrom je v běžné populaci vzácným typem epilepsie, navíc je v klinických zprávách publikováno několik případů, s vyšším podílem případů v ženské populaci.
Z epidemiologického hlediska je proto Ohtaharův syndrom považován za vzácné onemocnění, jeho prevalence se odhaduje na přibližně 0,2–4% všech dětských epilepsií.
Základní charakteristikou Ohtaharova syndromu je projevy záchvatů nebo epileptických záchvatů. Obvykle jsou záchvaty tonického typu, časté jsou však i myoklonické.
Obecně se příznaky epileptických záchvatů liší v závislosti na konkrétní etiologické příčině a individuálním klinickém průběhu, protože zatímco u některých lidí se zdá, že chybí na několik sekund, u jiných se projevují silné svalové záškuby.
Konkrétně v závislosti na strukturální expanzi a zdroji epileptického výboje lze epileptické jevy klasifikovat jako generalizované a ohniskové..
V případě Ohtaharova syndromu jsou záchvaty obvykle generalizované, to znamená, že abnormální výtok z neuronů postihuje všechny nebo dobrou část oblastí mozku.
Přestože existují různé typy generalizovaných záchvatů (absenční záchvaty, tonické, atonické, klonické, myklonické a tonicko-klonické záchvaty), nejčastější u Ohtaharova syndromu jsou tonické a myklonické:
- Tonické záchvaty: V tomto případě jsou epileptické záchvaty charakterizovány vývojem abnormálně zvýšeného svalového tonusu, to znamená výrazné svalové ztuhlosti, zejména na končetinách a zádech. Změna svalu v mnoha případech způsobí, že postižená osoba spadne.
- Myoklonické záchvaty: v tomto případě jsou epileptické záchvaty charakterizovány přítomností silných záškubů svalů v nohou a pažích.
Kromě toho je tento kardinální příznak charakterizován svou neřešitelnou povahou, ve většině případů klasické farmakologické a chirurgické přístupy používané při léčbě epilepsie obvykle u Ohtaharova syndromu nefungují..
Pokud jde o začátek klinických projevů Ohtaharova syndromu, epileptické záchvaty a záchvaty se obvykle začínají projevovat v raných fázích života..
Konkrétně se tonicko-myoklonické záchvaty obvykle začínají objevovat v prvních třech měsících života, v některých časných případech je to však patrné již za 10 dní po narození.
Po bezproblémovém porodu a normálním vývoji během prvních okamžiků života se záchvaty objevují akutně a náhle.
Tyto tonicko-myoklonické události tedy obvykle trvají přibližně 10 sekund a navíc se mohou objevit během spánkové fáze nebo během dne v bdělém stavu..
Za normálních okolností se klinický průběh Ohtaharova syndromu obvykle z důvodu zdravotních komplikací a vývoje závažného neurologického postižení (strukturního a funkčního) vyvíjí ve špatnou až špatnou lékařskou prognózu..
Většina lidí s Ohtaharovým syndromem umírá v raném dětství, v jiných případech se však tento zdravotní stav vyvine do Westova syndromu..
Děti s Ohtaharovým syndromem vykazují generalizovaný nedostatečný vývoj mozkových hemisfér v důsledku epileptických příhod a výtoků.
V důsledku toho bude u mnoha postižených docházet k významnému zpoždění psychomotorického vývoje, zvláště akcentovaného při získávání nových schopností a motorických dovedností v raném dětství..
Kromě toho, když se z této lékařské entity vyvine Westův syndrom, lze k výše uvedeným příznakům přidat některé z následujících:
- Infantilní křeče: trhnutí těla charakterizované celkovou flexí, ztuhlostí končetin a vyklenutím bederní oblasti.
- Hypsarytmie: Tato událost je definována jako zcela neuspořádaný mozkový elektrický výboj, charakterizovaný výboji pomalých vln, hrotů a ostrých vln s úplnou absencí hemisférické synchronizace.
- Regrese pohybových schopností: Kromě výrazných potíží se získáváním některých dovedností souvisejících se svalovou koordinací nebo ovládáním dobrovolných pohybů může při mnoha příležitostech dojít ke ztrátě schopnosti usmívat, držet hlavu, stát vzpřímeně nebo sedět..
- Svalová paralýza: je možný rozvoj diplegie, quadriplegie nebo tetraplegie.
- Mikrocefalie: vývoj zmenšeného obvodu hlavy ve srovnání s jedinci stejné věkové skupiny a pohlaví.
Etiologie epileptických encefalopatií, jako je Ohtaharův syndrom, je velmi různorodá.
Mezi nejběžnější však patří přítomnost nebo vývoj strukturálních změn v centrálním nervovém systému (CNS), metabolické patologie nebo genetické změny..
V případě genetických abnormalit prokázalo vyšetření některých případů přítomnost mutace v genu STXBP1 spojené s klinickým průběhem této patologie.
V současné době neexistuje žádný specifický test nebo test, který by jednoznačně indikoval jeho přítomnost, proto je diagnostický protokol sledovaný u Ohtaharova syndromu podobný jako u jiných typů epileptických poruch.
Na klinice lze kromě studia symptomů a charakteristik záchvatů a křečí použít některé doplňkové testy, jako je magnetická rezonance, elektroencefalografie, počítačová tomografie, neuropsychologické vyšetření nebo genetická studie..
Léčba použitá při Ohtaharově syndromu je založena hlavně na kombinaci různých léků používaných při jiných typech epileptických patologií.
Některé z přístupů tedy používají: fenobarbital, kyselinu valproovou, klonazepan, midazolan, vigabatrin, topiramát, mimo jiné..
Kromě toho se také testují další typy intervencí souvisejících s terapií steroidy, chirurgickým zákrokem, dietní terapií nebo léčbou metabolických poruch..
Většina z nich však nemá příznivý účinek při kontrole záchvatů a progresi onemocnění. V průběhu času se záchvaty opakují a jsou doprovázeny vážným narušením fyzického a kognitivního vývoje.

Zatím žádné komentáře