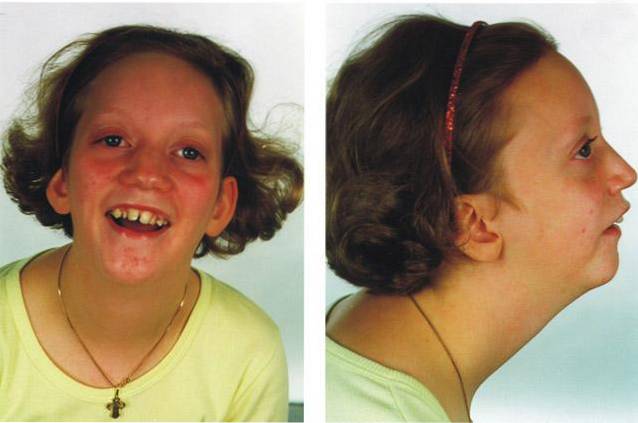
The Pierre Robinův syndrom Jde o poruchu genetického původu zařazenou do kraniofaciálních syndromů nebo patologií. Klinicky je charakterizována mikrognatií, glosoptózou, obstrukcí horních dýchacích cest a variabilní přítomností rozštěpu patra..
Pokud jde o etiologický původ této patologie, je Pierre-Robenův syndrom způsoben přítomností specifických mutací v genu SOX9, přičemž většina případů je diagnostikována.
Obecně tento syndrom vyvolává důležité zdravotní komplikace, včetně respiračního selhání, zažívacích zvířat nebo vývoje dalších kraniofaciálních malformací..
Na druhou stranu diagnóza Pierre-Robinova syndromu není obvykle potvrzena až do okamžiku narození; Kromě klinických nálezů je nezbytné provést různé radiologické testy k identifikaci kostních alterací..
V současné době neexistuje lék na syndrom Pierra Robina, nicméně k nápravě muskuloskeletálních abnormalit se často používají chirurgické přístupy. Kromě toho jsou důležité respirační a gastrointestinální úpravy, aby se zabránilo život ohrožujícím zdravotním komplikacím..
Rejstřík článků
Pierre Robinův syndrom je vrozená patologie, jejíž klinické nálezy jsou přítomny od okamžiku narození a navíc všechny jeho charakteristiky souvisejí s přítomností kraniofaciální malformace.
Dále v lékařské literatuře můžeme identifikovat různé termíny používané v kontextu Pierrova Robinova syndromu: Pierre Robinova choroba, Pierre Robinova malformace nebo Pierre Robinova sekvence.
Na specifické úrovni byl tento syndrom původně popsán v roce 1891 Meneradem a Lannelongue. V klinických zprávách popsali dva pacienty, jejichž klinický průběh byl charakterizován nedostatečným rozvojem struktury dolní čelisti, rozštěpem patra a lingválním posunem nebo zatažením.
Avšak až v roce 1923 Pierre Robin plně popsal klinické spektrum této patologie a své studie zaměřil na případ dítěte postiženého malformací dolní čelisti, abnormálně velkým jazykem a významnými dýchacími potížemi..
Navzdory skutečnosti, že se tato patologie zásadně odlišuje kraniofaciálními radiologickými nálezy, představuje vysokou mobilitu spojenou s lékařskými komplikacemi souvisejícími zejména se srdečním selháním a problémy s krmením..
Konkrétně má syndrom Pierre Robin vysokou úmrtnost spojenou s obstrukcí dýchacích cest, neurologickými abnormalitami nebo srdečními abnormalitami.
Na druhou stranu mnoho autorů dává přednost této patologii pouze jako Pierrova posloupnost, protože právě mandibulární anomálie mají tendenci produkovat zbytek typických znaků a symptomů..
Prevalence syndromu Pierra Robina se odhaduje na přibližně jeden případ na každých 8 500 živě narozených dětí, z nichž více než 80% diagnostikovaných případů je spojeno s dalšími zdravotními komplikacemi a specifickými syndromy.
Na druhé straně je v případě Spojených států výskyt syndromu Pierra Robina 1 případ na 3120 narozených každý rok.
V současné době nebyl zjištěn ani rozdílný výskyt syndromu Pierra Robina spojený se sexem, geografií nebo konkrétními etnickými a rasovými skupinami..
Jak jsme již dříve zdůraznili, syndrom Pierra Robina představuje jednu z kraniofaciálních patologií s vysokou pravděpodobností úmrtnosti. Ve Spojených státech přibližně 16,6% postižených zemře na vývoj zdravotních komplikací.
V pořadí výskytu jsou nejčastějšími sekundárními medicínskými patologiemi: srdeční anomálie (39%), změny v centrálním nervovém systému (33%) a anomálie v jiných orgánech (24%).
Sekvence Pierra Robina se odlišuje od ostatních typů mozkových cév patologií přítomností tří základních klinických znaků: mikrognathie, glosoptóza a rozštěp patra:
Termínem micrognathia označujeme přítomnost patologické změny ve vývoji struktury dolní čelisti, konkrétně konečný tvar představuje zmenšenou velikost ve srovnání s úrovní očekávanou pro úroveň vývoje postižené osoby..
V důsledku toho neúplný vývoj této kraniofaciální struktury způsobí širokou škálu změn, které všechny souvisejí s přítomností malformací, které postihují ústa a obličej..
Micrognathia je lékařský znak přítomný přibližně u 91% lidí postižených syndromem Pierra Robina..
Termínem glosoptóza označujeme přítomnost abnormálního zatažení polohy jazyka v ústní struktuře, konkrétně mají jazyky tendenci být umístěny více dozadu než obvykle v důsledku mikrofotografie a zmenšení objemu ústní dutina.
Abnormality týkající se polohy a struktury jazyka mohou způsobit závažné problémy s krmením, které mohou vést k vážným zdravotním stavům..
Kromě toho je v jiných případech také možné identifikovat abnormálně velký jazyk (makroglosii), který mimo jiné ztěžuje dýchání, žvýkání nebo produkci funkčního jazyka..
Glosoptóza je navíc jedním z nejčastějších klinických příznaků u syndromu Pierra Robina, pozorovaného u přibližně 70–85% diagnostikovaných případů. Zatímco makroglosii lze pozorovat v menším procentu, asi u 10–15% postižených jedinců.
Tento termín označuje přítomnost malformace v palatálních oblastech nebo bukální střeše, to znamená, že lze pozorovat přítomnost trhlin nebo otvorů spojených s neúplným vývojem dolní čelisti..
Stejně jako ostatní klinické nálezy, rozštěp patra způsobí významné změny v krmení.
Kromě těchto známek a příznaků je také možné identifikovat další typy změn, včetně:
- Malformace nosu.
- Poruchy oka.
- Muskuloskeletální změny a malformace, hlavně související s vývojem oligodaktylie (snížení počtu prstů, méně než 5 v rukou nebo nohou), klinodaktylie (příčná odchylka polohy prstů), polydaktylie (zvýšený počet prstů), hypermobilita v kloubech (abnormálně přehnané zvýšení pohyblivosti kloubů), dysplazie ve falangách (falangy se špatným nebo neúplným vývojem kostí) nebo syndaktylie (fúze několika prstů).
- Další změny: je také možné identifikovat malformace ve struktuře končetin nebo v páteři.
Kromě výše popsaných zdravotních funkcí se mohou objevit i další související s různými systémy:
Srdeční změny představují jednu z lékařských komplikací s největším dopadem na zdraví jedince a představují významná rizika pro jejich přežití. Známky a příznaky související s kardiovaskulárním systémem jsou však obvykle léčitelné pomocí farmakologických a / nebo chirurgických přístupů..
Mezi nejčastější srdeční abnormality patří srdeční stenóza, přetrvávající foramen ovale, změněné septické tepny nebo hypertenze..
Genetický původ syndromu Pierra Robina může také znamenat vývoj různých neurologických změn, zejména souvisejících s přítomností abnormalit v centrálním nervovém systému (CNS)..
Mezi neurologické poruchy nejčastěji spojené se syndromem Pierra Robina tedy patří hydrocefalus, Chiariho malformace, epileptické epizody nebo zpoždění v získávání psychomotorických dovedností..
Respirační změny jsou jedním z nejdůležitějších rysů, protože mohou způsobit smrt pacienta v důsledku respiračního selhání a poškození mozku v důsledku nedostatku kyslíku v nervových oblastech.
V mnoha případech jsou tedy nutné chirurgické korekce k uvolnění dýchacích cest, zejména korekce mandibulární dysplazie nebo polohy jazyka..
Stejně jako v případě respiračních poruch jsou problémy s krmením zásadně odvozeny od malformací dolní čelisti.
Proto je od narození nezbytné identifikovat ty abnormality, které ztěžují krmení, aby bylo možné je napravit a snížit tak pravděpodobnost vzniku zdravotních stavů souvisejících s podvýživou..
Syndrom nebo sekvence Pierra Robina má genetický etiologický původ, spojený se změnami v genu SOX9. Ačkoli byla tato anomálie identifikována ve většině ojedinělých případů syndromu Pierra Robina, některé jeho klinické charakteristiky mohou souviset s jinými typy mutací genetického původu..
Specificky má gen SOX9 základní úlohu při poskytování nezbytných biochemických pokynů pro produkci proteinu podílejícího se na vývoji a tvorbě různých tkání a orgánů během vývoje plodu..
Současný výzkum navíc naznačuje, že protein SOX9 může regulovat aktivitu jiných typů genů, zejména těch, které se podílejí na vývoji kosterní, a tedy mandibulární struktury..
Výsledkem je, že genetické změny zabraňují správnému morfologickému vývoji určitých struktur, a proto se objevují kardinální klinické nálezy: mykognatie, glosoptóza a rozštěp patra..
V mnoha případech lze kraniofaciální strukturální malformace identifikovat během těhotenství pomocí ultrazvukových vyšetření, i když případy jsou vzácné.
V tomto smyslu je častější podezření na syndrom Pierra Robina v postnatální nebo infantilní fázi. U většiny postižených jsou strukturální příznaky výrazně patrné, takže diagnóza je potvrzena radiologickými testy spolu s fyzikálním vyšetřením..
V druhém případě je však nutné předem provést respirační studii a následně radiologickou studii, aby se zjistila přítomnost tohoto syndromu..
Kromě toho je dalším základním aspektem v diagnostice této patologie zkoumání dalších oblastí, zejména srdečního a nervového systému, protože se mohou objevit jiné typy život ohrožujících anomálií.
A konečně, diagnostický zásah může zahrnovat individuální a rodinnou genetickou studii k identifikaci možných genetických asociací..
Typická léčba syndromu Pierra Robina je založena na chirurgických zákrocích k nápravě kraniofaciálních malformací:
- Tracheotomie.
- Patrový rázštep uzavření.
- Prodloužení čelisti.
- Rozptýlení kostí.
- Jazyková fixace.
Kromě toho se k léčbě srdečních patologií, epileptických epizod a dalších neurologických příhod používají také další farmakologické přístupy..
Kromě toho mají postižení lidé potíže spojené s produkcí jazyka, a proto je v mnoha případech zásadní časný logopedický přístup..
Základním cílem je zavést efektivní komunikační metodu prostřednictvím zbytkových kapacit a následně stimulace získávání nových dovedností..



Zatím žádné komentáře