
The Waardenburgův syndrom (SW) jedná se o patologii genetického původu klasifikovanou jako typ neuropatie. Jeho klinické vlastnosti jsou definovány přítomností hluchoty nebo ztráty sluchu, abnormální pigmentací očí, vlasů nebo kůže a různými změnami obličeje.
Tato patologie je charakterizována svou širokou symptomatickou variabilitou, a proto se rozlišuje několik typů: typ I, typ II, typ III (Klein-Waardenburgův syndrom nebo psudo Waardenburg) a typ IV.

Na etiologické úrovni má Waardenburgův syndrom autozomálně dominantní vzor dědičnosti. Obvykle je spojena se specifickými mutacemi v genech EDN3, EDNRB, PAX3, SOX10, SNAI2 a MIT..
Diagnóza je stanovena na základě různých hlavních a vedlejších klinických kritérií. Je však nutné provést různé doplňkové laboratorní testy. Neexistuje žádná specifická léčba nebo léčba Waardenburgova syndromu..
Intervence s touto patologií se obvykle zaměřuje na léčbu poruch sluchu (chirurgické zákroky, kochleární implantáty atd.), Logopedii a neuropsychologickou rehabilitaci i psychologickou.
Rejstřík článků
Tento syndrom původně popsal nizozemský genetik a oftalmolog Petrus Johannes Waardenburg v roce 1848. Ve své klinické zprávě poukázal na hlavní klinické charakteristiky:
Následné analýzy identifikovaly velkou klinickou variabilitu Waardenburova syndromu. Kromě toho Mckusick spojoval tento syndrom s dalšími podobnými klinickými průběhy, jako je Hirschsprungova choroba..
V současné době je považována za vzácnou patologii, která se vyskytuje s různým stupněm poruchy sluchu, která může způsobit významné změny v učení a pozdějším vývoji postižené osoby.
Prognóza Waardenburgova syndromu je příznivá, i když může být spojena s významnou morbiditou a mortalitou související s lékařskými komplikacemi, zejména střevními.
Waardenburgův syndrom je vrozená genetická porucha, jejíž příznaky a příznaky se u postižených obvykle velmi liší..
Mezi nejčastější rysy patří výrazné abnormality obličeje, změněná pigmentace kůže, očí nebo vlasů a hluchota..
V lékařské literatuře je tento syndrom často považován za typ genodermatózy nebo neuropatie. Termín genodermatóza označuje širokou škálu onemocnění charakterizovaných přítomností kožních abnormalit a změnami genetického původu..
Na druhé straně termín neuropatie označuje skupinu patologií odvozených z vývoje anomálií a defektních procesů během migrace a diferenciace buněk nervové lišty během těhotenství..
Nervový hřeben je embryonální struktura tvořená širokou sadou nediferencovaných buněk, jejichž vývoj povede k vytvoření kranio-obličejové struktury a neuronálních a gliových buněk, které budou tvořit velkou část nervového systému..
Mezi 8. a 10. týdnem těhotenství obvykle začíná proces migrace buněk, které tvoří neurální lištu. Pokud do tohoto procesu zasahují různé patologické faktory nebo genetické abnormality, mohou se objevit významné kognitivní a / nebo fyzické abnormality, jako je Waardenburgův syndrom..
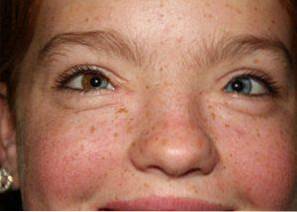
Prevalence Waardenburova syndromu se odhaduje na 1 případ na každých 40 000 lidí na celém světě. Od svého objevu bylo v lékařské a experimentální literatuře popsáno asi 1400 různých případů..
Zdá se, že to ovlivňuje muže i ženy stejně. Nebyly identifikovány žádné asociace s geografickými regiony nebo konkrétními etnickými a rasovými skupinami.
Waardenbugův syndrom představuje 2–5% všech diagnostikovaných případů vrozené ztráty sluchu.
Ačkoli byly identifikovány různé klinické průběhy, nejčastější jsou typy I a II. Typ III a IV jsou vzácné.

Waardenburgův syndrom se vyznačuje třemi základními změnami: kraniofaciálními změnami, pigmentovými abnormalitami a hluchotou:
Dalším z hlavních lékařských nálezů Waardenburgova syndromu je ztráta sluchové schopnosti a ostrosti. Nejběžnější je identifikovat u postižených různý stupeň hluchoty nebo senzorineurální ztráty sluchu.
Termín senzorineurální ztráta sluchu označuje ztrátu sluchové schopnosti způsobenou vnitřními poraněními souvisejícími s nervovými zakončeními, která vedou sluchové informace z vnitřního ucha do mozkových center..

Waardenburgův syndrom je klasifikován do 4 základních typů na základě klinického průběhu a konkrétních příznaků, které jsou přítomny u postižených lidí:
Waardenbuugův syndrom má vrozený původ spojený s různými genetickými změnami.
Analýza případů umožnila lokalizovat tyto anomálie v genech: EDN3, EDNRB, PAX3, SOX10, SNAI2 a MIT.
Zdá se, že tato sada genů se podílí na vývoji a tvorbě různých typů buněk, včetně těch, které jsou odpovědné za produkci melanocytů.
Melanocyty jsou odpovědné za tvorbu melaninu, pigmentu, který přispívá ke zbarvení očí, vlasů nebo pokožky.
V závislosti na různých klinických kurzech můžeme identifikovat různé genetické změny:
Jak jsme zdůraznili v původním popisu, diagnóza Waardenbugova syndromu je stanovena na základě několika hlavních a vedlejších kritérií:
Pro stanovení konečné diagnózy je nezbytné identifikovat přítomnost dvou hlavních kritérií nebo alespoň jednoho hlavního a dvou vedlejších. Kromě toho je nutné použít některé doplňkové testy: biopsie, audiometrie nebo genetické testy.
Léčba Waardenbugova syndromu neexistuje, i když lze použít symptomatické přístupy..
Léčba nejběžnějších známek a příznaků obvykle vyžaduje lékařský zásah dermatologů a oftalmologů..
Na druhou stranu v případě léčby senzorineurální hluchoty lze provést kochleární implantát doprovázený logopedickou a neuropsychologickou intervencí..



Zatím žádné komentáře